



通过双电子转移氧还原反应(2e−-ORR)合成电化学双氧水(H2O2)有望取代高能耗和二氧化碳排放密集型的蒽醌工艺。碳基质(Co-N-C)催化剂中的氮配位单原子Co已被公认为潜在的催化剂候选者,且它们的性能在很大程度上取决于Co活性位点周围的局部原子结构。然而精确地构建催化剂的原子结构仍然极具挑战性,在很大程度上限制了高活性催化剂的设计及其活性的提高。因此实现催化剂原子结构的精确控制对于实现高性能催化剂至关重要。然而,对于传统的基于热解的合成方法来说,这仍然是一项具有挑战性的任务。多相分子催化剂(HMCs)可以将分子催化的原子结构明确地引入非均相催化剂,无疑是一种可行的解决方案。而钴卟啉具有类似于ORR活性Co-N4的位点,并可以通过范德华作用吸附在催化惰性碳纳米管(CNT)表面,形成具有明确和可调原子结构的HMCs。同时钴卟啉原子结构可以通过严格的有机合成调控,在meso和β位点的H原子可被替换为不同官能来实现不同的功能。具有各种meso取代基的钴卟啉已被广泛研究,其不同的ORR性能主要归因于取代基的空间位阻或辅助因子效应。β-取代基可以修饰卟啉π电子体系并调整Co电子性能,这为精确优化Co位点H2O2合成性能提供了绝佳的机会。
近日,澳大利亚悉尼大学化学及生物化工学院魏力、陈元老师团队和日本东北大学先进材料研究中心李昊老师(共同通讯作者)团队通过第一性原理计算发现,卟啉β位取代基和碳载体可以协同调节Co中心的电子结构和催化活性。团队预测八氟取代的催化剂为最佳催化剂,并通过实验进一步验证该猜想。实验结果显示,在酸性电解质及200毫伏过电位下,八氟取代的非均相分子催化剂可以实现>94%的H2O2选择性(摩尔分数)和3.51每秒的周转频率(TOF)。在双电极电解槽中,它可以达到最高为10.76 molH2O2 gcat−1h−1的H2O2生产率,可作为水处理和化工生产中使用纯H2O2的解决方案。研究成果以“Heterogeneous molecular Co–N–C catalysts for efficient electrochemical H2O2 synthesis”为题发表于著名能源和环境科学综合期刊《Energy & Environmental Science》(IF 39.714)上,并被选为最新一期的内封面文章。

研究亮点
Energy & Environmental Science
1. 催化剂性质和活性的理论预测
首先研究了β取代的卟啉(H2PorX,X=H,Et,Br和F)以及对应钴卟啉的电子结构,并通过对比发现β取代基可改变钴的性质。此外,采用卟啉的分子模型和石墨平面构建的HMC原子模型发现卟啉π电子与石墨烯的重叠pz轨道会导致两种组分之间产生明显的电荷转移现象,并因此,碳纳米管衬底可以与β取代基协同调整Co性质。通过分析Co中心与不同ORR中间体之间的相互作用,预测了这些催化剂模型的ORR活性。在取代基类型方面,计算结果定性预测了ORR活性趋势:H≈F>Et>Br,H2O2的选择性趋势:Br>F>H>Et。
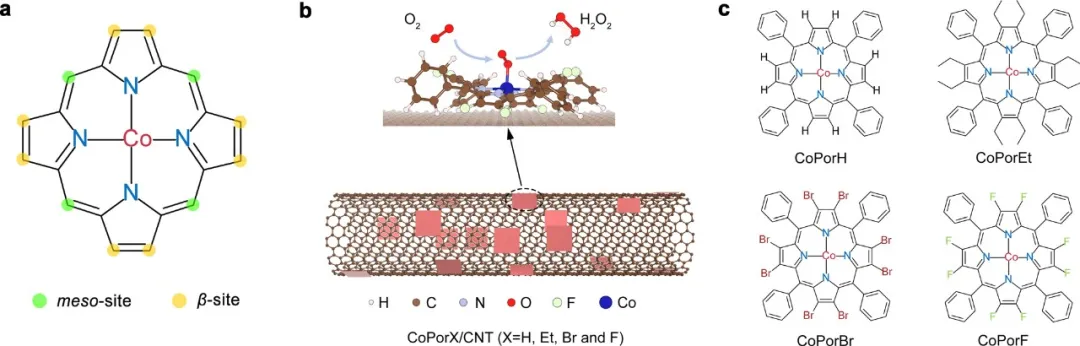
图1 多相分子催化剂的合成。(a)卟啉的化学结构和两类取代位点。(b)用于H2O2电合成的HMC的制备示意图。(c)本研究的钴卟啉的分子结构。
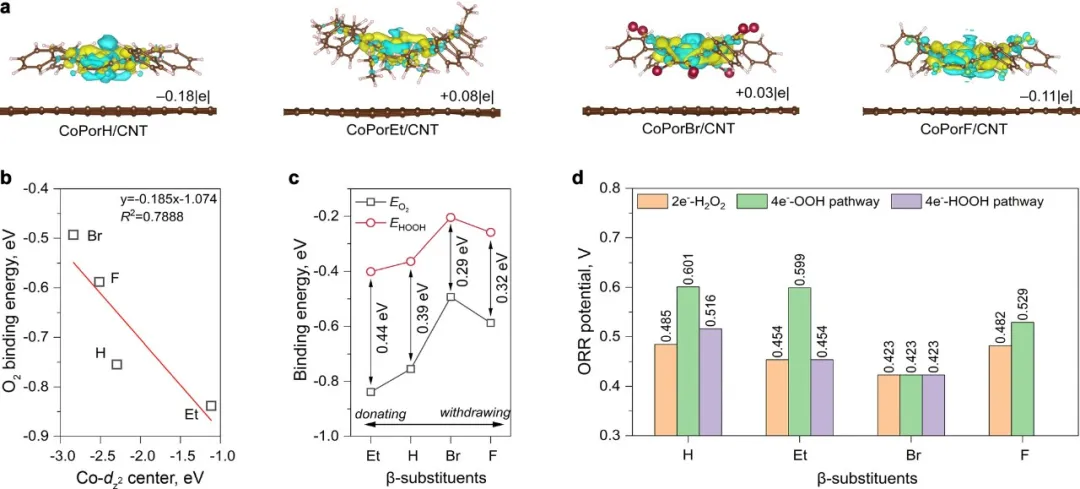
图2 HMC模型的理论预测。(a)通过向石墨烯表面添加卟啉分子引起的各种HMC模型的差分电荷密度(黄色和青色等值表面分别表示电荷积累和消耗,等值值为0.002 Å−3).(b)计算出的Co-dz2轨道中心和*O2结合能(EO2)之间的关系。(c)β取代基与中间体*O2和*HOOH(EHOOH)的计算结合能之间的关系。(d)沿不同反应途径的理论ORR电位。
2, 非均相分子催化剂的合成与表征
基于此前的理论原理指导,相应地合成了β取代的卟啉和钴卟啉。在DMF中,合成的钴卟啉通过范德华力吸附在CNT表面,然后再经过滤和洗涤获得非均相分子催化剂(HMC)。在HAADF-STEM图中,高度分散的亮点对应于卟啉分子中的单个钴原子,没有观察到卟啉聚集体。通过XANES发现不同β取代卟啉的Co价态接近2+,略有差异。此外通过Co–N键长与卟啉分子变形参数之间的相关性可以发现,随着β取代基尺寸和/或分子量更大,卟啉分子畸变得更加显著。由XPS发现,钴卟啉在碳纳米管衬底上吸附后,所有钴卟啉中Co的峰值均发生一定偏移。CoPorH/CNT和CoPorF/CNT将损失0.08-0.11|e|,将有他们的2p3/2峰转向更高的结合能,而CoPorEt/CNT和CoPorBr/CNT表现出相反的趋势,这一现象确认吸附的卟啉分子与CNT底物之间的强电子相互作用。
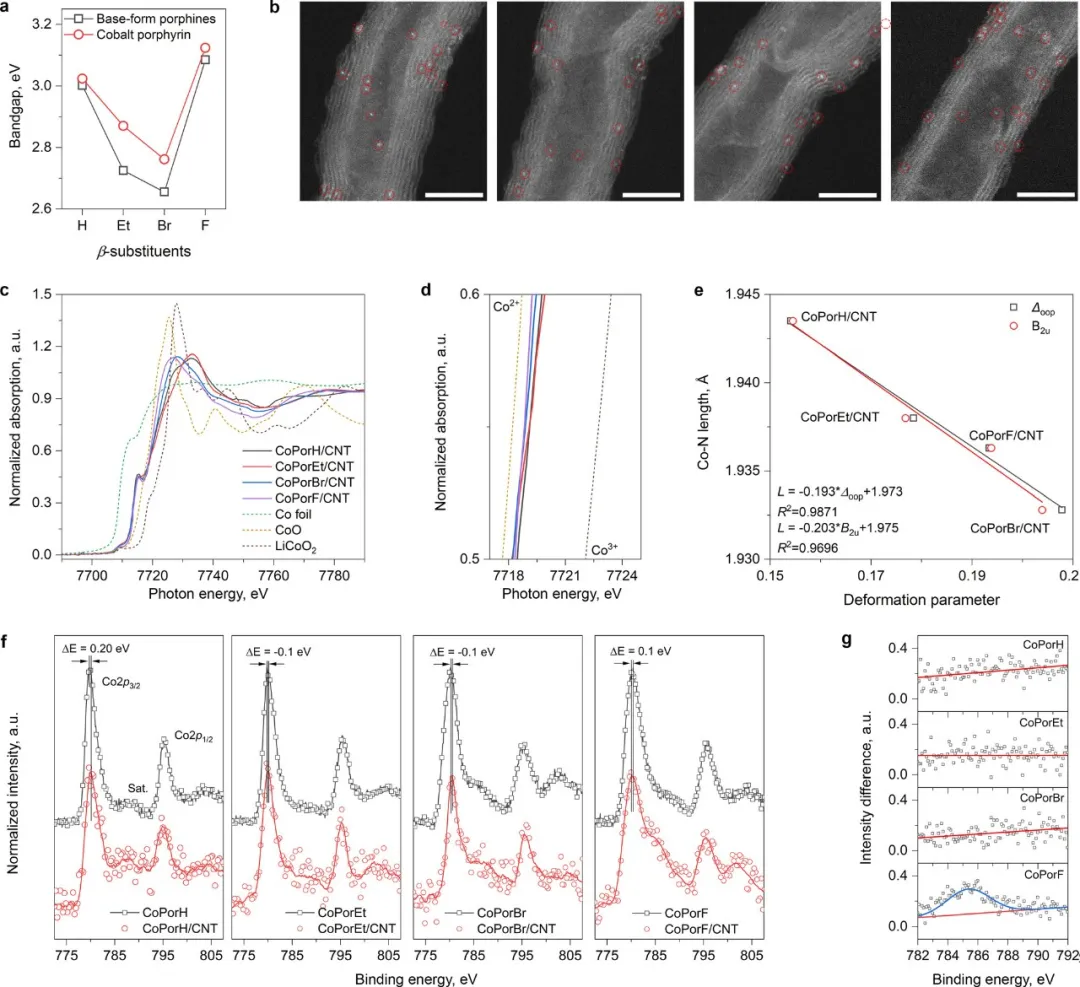
图3 分子和HMC的表征。(a)根据CH2Cl2中紫外-可见吸收光谱确定的β取代卟啉和钴卟啉的带隙。(b) 催化剂的HAADF-STEM图像,比例尺为5nm。左起依次为CoPorH/CNT、CoPorEt/CNT、CoPorBr/CNT和CoPorF/CNT。(c)HMC和对照组钴箔、CoO 和 LiCoO2的Co K-edge XANES光谱。(d)样品吸收边放大图。(e)Co-N键长与卟啉分子变形参数之间的相关性。(f)卟啉分子在碳纳米管衬底上吸附前后的高分辨率Co 2p XPS光谱。(g)Co 2p XPS光谱在2p3/2卫星区域处的差异。
3, 三电极测试中的ORR性能
在RRDE上以0.01 mg cm2进行的HMC ORR性能测试。对比0.1M HClO4下CNT的LSV曲线,所有HMCs均在0.7 V存在明显的电流,表明HMC的性能均来自Co位点。所有HMCs的过电位反映出催化活性趋势与此前的预测相同:H≈F>Et>Br。进一步计算HMCs的H2O2选择性,选择性大小关系为Br>F>H>Et,该结果与此前作者采用的描述符(*O2-*HOOH)预测趋势一致。通过计算HMCs的周转频率TOF,作者发现CoPorF/CNT在200 mV的过电位下可以提供3.51±0.06 s−1,优于其他HMC和最近报道的原子钴和其他单原子金属催化剂。此外,HMCs还在碱性电解质中进行了ORR测试,结果显示所有HMCs的ORR电流均得到了改善,但选择性有所下降。作者发现两者的性能差异与HMC中Co中心在氧中间体的吸附前后的偶极矩变化单调相关。
图4 通过三电极测量获得ORR性能。O2饱和0.1 M HClO4电解质中获得的(a)LSV曲线和(b)相应的H2O2法拉第效率(扫描速率:5 mV s−1,转速:1600 rpm,无iR校正)。(c)HMC与最近报告的一些催化剂TOF对比。实线/符号:来自酸性电解质;虚线/开放符号:来自碱性电解质。参考单原子催化剂的TOF值通过直接引用或根据其金属含量和催化剂负载量使用与本工作相同的方法计算。与HMC一样,这些催化剂中的所有金属都被认为是活性位点。对于碳基催化剂,活性位点的数量是根据–C-O-C-构型中的氧含量估计的,该构型被认为是关键的ORR活性位点。O2饱和0.1 M KOH电解质中获得的(d)LSV曲线和(e)相应的H2O2法拉第效率。
4, H2O2双电极电解槽的生产性能
作者利用双电极流通池电解槽中对HMC的H2O2生产性能经行了测试。电解槽的电流-电压响应趋势与RRDE测试结果一致,且CoPorF/CNT表现出最优的性能。在50 mA cm−2下,CoPorF/CNT可以在在12 mL h−1的去离子水流动速度下持续生成9300ppm的H2O2溶液,通过降低DI水流动速率可以实现18200ppm的H2O2溶液。作者进一步探究了HMCs失活的原因。稳定性测试后样品的XANES和ICP-AES揭示CoPorEt/CNT和CoPorBr/CMT失活的原因可能是Co的在卟啉分子上的脱附。计算结果显示Et和Br取代基的较大尺寸可能导致更为严重的卟啉分子变形,中间体的吸附导致两者模型的核心变形程度进一步加剧。相反,CoPorF/CNT模型在过程中变得扁平化,结构完整性得到改善,这表明β取代基也可以控制Co-N4活性位点在电化学H2O2合成中的稳定性。最后作者使用地表水样本中去除TOC和选择性苯酚羟基化反应测试CoPorF/CNT产生H2O2的实用性。
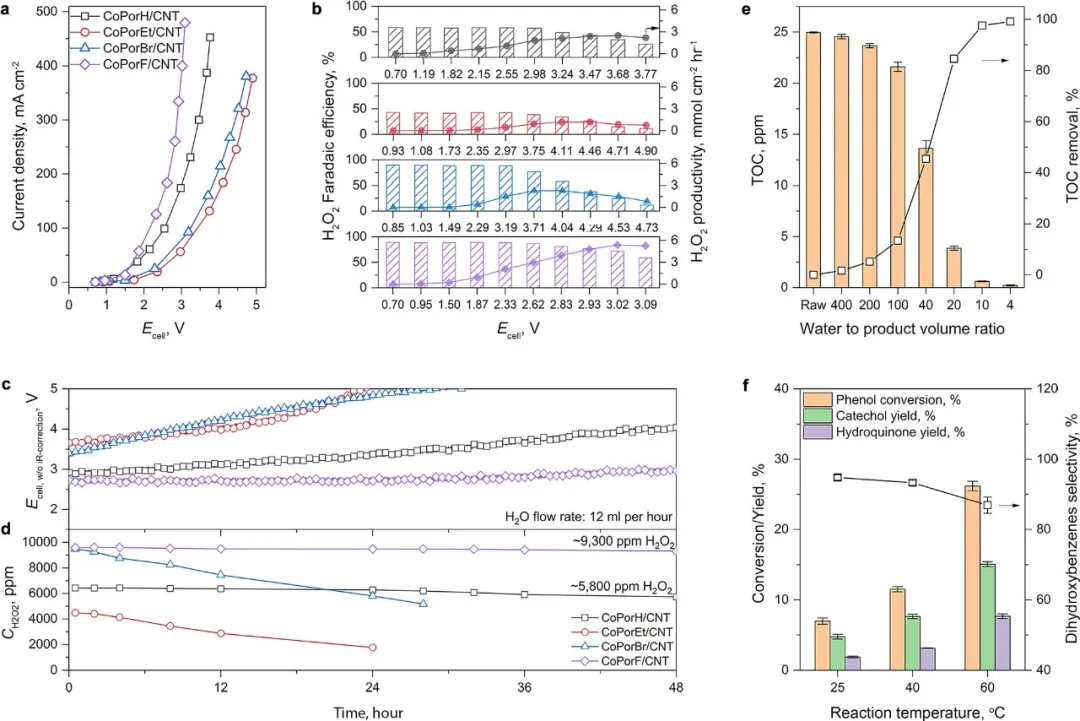
图5 流通相电解槽中的ORR性能以及生产的H2O2溶液的应用。(a)80%-iR校正下的电压-电流响应。(b)在不同条件下获得的H2O2法拉第效率和生产率。(c)计时安培性能和(d)不同催化剂在50 mA cm−2和水流速为12 mL h−1时的H2O2浓度。采用CoPorF/CNT催化剂生产H2O2的应用。(e)从地下水样本中去除TOC。(f)苯酚羟基化反应性能。
通讯作者
Energy & Environmental Science
陈元 教授
陈元,悉尼大学化学和生物分子工程学院教授。本科、硕士毕业于清华大学,博士毕业于耶鲁大学。2005-2015年任南洋理工大学助理教授、副教授。2015年起任悉尼大学教授。2017年当选澳大利亚皇家化学会会士,2018年当选英国皇家化学会会士。目前还担任国际期刊Carbon和Journal of Alloys and Compounds编辑。主要研究方向是碳纳米材料的可控合成、组装和其在能源和环境方向的应用。
课题组网站:https://yuanchenlab.org/
李昊 副教授
李昊,副教授,2022年加入日本东北大学(Tohoku University)材料科学高等研究所(AIMR),作为课题组PI组建“数字催化研究课题组”,从事材料设计与计算、机器学习研究。于2019年博士毕业于美国德克萨斯大学奥斯汀分校(师从Graeme Henkelman教授)。2020-2022年初于丹麦科技大学从事博士后工作,师从现代催化理论鼻祖、美国工程院院士Jens K Nørskov教授。已在Nature Catal.、Nature Commun.、JACS、Adv. Mater.、ACS Catal. 、Energy & Environ. Sci..等杂志发表论文120余篇。曾获荣誉包括“表面科学青年学者”(美国化学年会2022)、被美国化学工程师协会(AIChE)评为年度最佳基础学科研究、被英国皇家化学会(RSC)旗下杂志评为年度杰出青年材料化学学者。曾被普林斯顿、苏黎世联邦理工、慕尼黑工大、澳洲国立大学、昆士兰大学等学府及会议邀请作研究报告20余次。
欢迎联合培养/访问学生以及访问学者莅临合作。也欢迎依托李昊课题组申请公派留学生项目(CSC)。有兴趣欢迎将个人简历发至李昊教授的邮箱。
邮箱:li.hao.b8@tohoku.ac.jp
魏力 讲师
魏力,悉尼大学化学和生物分子工程学院讲师。分别于清华大学和南洋理工大学获得本科和博士学位。于2017年加入悉尼大学。目前也受澳大利亚研究基金会未来学者基金资助。主要研究方向是基于碳及金属氧化的电催化剂在氢气,氧气,二氧化碳相关催化转化中的应用及器件研发。
文章信息
Energy & Environmental Science
Chang Liu, Zixun Yu, Fangxin She, Jiaxiang Chen, Fangzhou Liu, Jiangtao Qu, Julie M. Cairney, Chongchong Wu, Kailong Liu, Weijie Yang, Huiling Zheng, Yuan Chen*, Hao Li* and Li Wei*. Heterogeneous molecular Co–N–C catalysts for efficient electrochemical H2O2 synthesis. Energy Environ. Sci., 2023, 16, 446-459.

